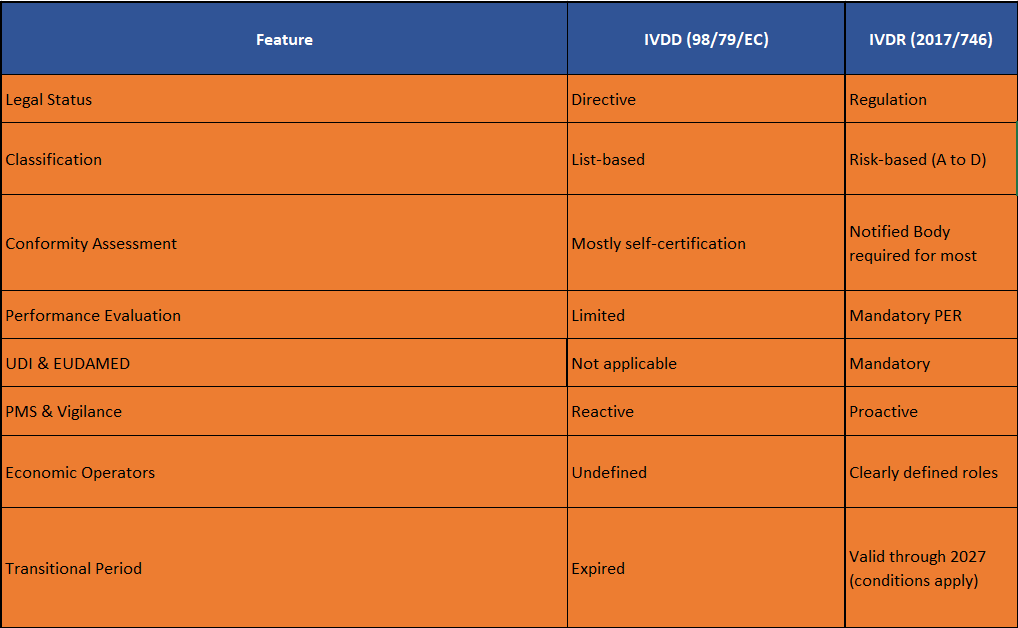
Table of Contents
When the In Vitro Diagnostic Medical Devices Regulation (EU) 2017/746 (IVDR) officially replaced the In Vitro Diagnostic Directive 98/79/EC (IVDD) on May 26, 2022, it marked a fundamental shift, not just a regulatory upgrade. IVDR introduces stricter controls, deeper oversight, and expanded responsibilities across the IVD supply chain.
Let’s break down what’s changed and how it impacts your IVD compliance strategy.
IVDR Is a Regulation — Not a Directive
The IVDD was a directive—each EU country could interpret and transpose it into national law. The IVDR, by contrast, is a regulation. That means it’s directly applicable across all EU Member States, eliminating loopholes and inconsistencies.
Impact: Uniform rules across the EU. No more fragmented compliance frameworks.
Device Classification: From List-Based to Risk-Based
IVDD Classification:
- Mostly self-certified
- Few high-risk devices require Notified Body oversight
- Classification was largely list-based (Annex II & self-test devices)
IVDR Classification (Annex VIII): Now, IVDs are classified by risk to patient and public health:
- Class A: Low individual/public risk (e.g., lab instruments)
- Class B: Moderate risk (e.g., pregnancy tests)
- Class C: High risk (e.g., cancer diagnostics)
- Class D: Highest risk (e.g., HIV, blood grouping tests)
Impact: Under IVDR, up to 85% of IVDs now require Notified Body involvement, compared to 15% under IVDD.
Performance Evaluation Requirements
Under IVDD: Limited clinical evidence requirements for many devices.
Under IVDR: Mandatory performance evaluation for every IVD:
- Scientific Validity
- Analytical Performance
- Clinical Performance
All must be documented in a Performance Evaluation Report (PER), updated throughout the lifecycle.
Impact: Significant increase in data generation, documentation, and ongoing updates—even for legacy products.
General Safety and Performance Requirements (GSPR)
IVDD → Essential Requirements (Annex I) IVDR → General Safety and Performance Requirements (Annex I)
Expanded and more specific requirements under IVDR include:
- Chemical, biological, and physical properties
- Infection and contamination control
- Measurement accuracy
- UDI traceability
- Labeling clarity in all EU languages
- Risk control and lifecycle management
Impact: More documentation. Broader supplier traceability. New design, testing, and validation checkpoints.
Conformity Assessment: New Routes and Oversight
Under IVDD:
- Most manufacturers could self-certify (Annex III)
- Few needed Notified Body review
Under IVDR: Conformity assessment depends on classification and includes:
- Notified Body audit of Quality Management System (QMS)
- Product sampling for Class C/D
- Performance Evaluation File (PEF) and PER review
- Use of EU certificates of conformity
Impact: Drastic reduction in self-certification. Complex, audit-ready documentation required for most devices.
Unique Device Identification (UDI) and EUDAMED
Not covered under IVDD.
Under IVDR:
- Devices require a UDI to ensure traceability
- Manufacturers, Authorized Reps, Importers must register in EUDAMED
- Devices must be registered in EUDAMED with UDI-DI and basic UDI-DI elements
Impact: Mandatory digital traceability for all IVDs across the EU market.
Post-Market Surveillance (PMS) & Vigilance
IVDD: Focused on reactive reporting.
IVDR: Requires a proactive PMS plan, including:
- PMS reporting procedures
- Trend reporting
- Vigilance reporting (15-day deadline for serious incidents)
- Periodic Safety Update Report (PSUR) for Class C & D devices
- Post-Market Performance Follow-up (PMPF) as a continuous process
Impact: Constant lifecycle monitoring is now legally required—not optional.
New Economic Operator Responsibilities
Under IVDD, obligations were mostly on the manufacturer.
Under IVDR, four economic operators are clearly defined:
- Manufacturer
- Authorized Representative
- Importer
- Distributor
Each must verify compliance, maintain documentation, and report incidents.
Impact: Compliance is now a shared responsibility—and your supply chain is under scrutiny.
Transitional Provisions & Timelines
The EU has staggered IVDR implementation deadlines for legacy devices:
- Class D: May 26, 2025
- Class C: May 26, 2026
- Class B & Class A (sterile): May 26, 2027
Only applicable if:
- Devices were placed under IVDD with valid certificates
- No significant design or intended use changes
- Devices continue to meet IVDD and IVDR PMS/vigilance obligations
Impact: Manufacturers must work backward from deadlines and prepare new technical documentation now.
Summary: IVDR vs IVDD
How Acquis Simplifies Your IVDR Transition
IVDR isn’t just paperwork—it’s a supply chain transformation. Acquis helps you stay ahead with:
- Automated GSPR & PER workflows
- UDI/EUDAMED integration
- Notified Body audit readiness
- Supplier declaration traceability
- Real-time PMS & vigilance tools Request a free IVDR compliance review to find out how ready your IVD operations really are.